用于检测伴侣未知的基因融合的方法与流程
用于检测伴侣未知的基因融合的方法
1.相关申请的交叉引用
2.本技术根据35u.s.c.119(e)要求于2019年3月22日提交的美国临时申请第62/822,429号的权益。前述申请的全部内容以引用的方式并入本文中。
技术领域
3.本技术总体上涉及用于检测基因融合的方法、系统和计算机可读介质,并且更具体地涉及基于使用下一代测序技术对驱动基因中的外显子
‑
外显子连接处对靶向rna测序的基因融合进行的伴侣未知的检测。
技术实现要素:
4.由像alk、ret、ntrk1等驱动基因中的染色体重新布置事件产生的基因融合转录物已成为癌症诊断和靶向疗法选择的重要生物标志物。根据各个示例性实施例,提供了用于基于驱动基因中的一些或所有外显子
‑
外显子连接处的靶向rna测序对基因融合进行检测的方法。测量每个外显子
‑
外显子连接处的表达并且检测表达不平衡模式可以预测样品中涉及所述驱动基因的基因融合事件。基因融合事件可以以伴侣未知的方式检测,即不使用对特定融合伴侣基因或特定断点信息的任何先验知识。所述方法可以检测涉及测试样品中的任何靶向驱动基因的融合,并且可以预测驱动基因内被鉴定为对于融合为阳性的近似断点位置。基因融合事件是与置信度评分和p值一起检测和报告的。基于来自一组正常样品的读段数据构建的rna基线可提高融合检测的稳健性和准确性。本文所述的方法的结果可以与如所靶向融合同种型测序等融合检测的其它方法的结果一起报告。
5.基于表达失衡对基因融合进行检测由于各种因素,如驱动基因基于样品类型、组织类型、条形码多路复用和肿瘤含量的rna表达的可变性而具有挑战性。本文描述的方法借助于通过将每基因多个扩增子放置在具体模式中将基因融合产物中的不平衡的表达特征建模为覆盖率模式检测问题、对驱动基因中的扩增子的覆盖率表达值进行归一化,应用利用依据一组正常样品计算的基线进行的基因特异性校正并且计算不平衡评分和p值来解决这些挑战中的一些挑战。
6.根据示例性实施例,提供了一种用于检测基因融合的方法,所述方法包括:(a)在引物池存在的情况下对核酸样品进行扩增以产生多个扩增子,所述引物池包含靶向驱动基因的多个外显子
‑
外显子连接处的引物,其中所述扩增子与所靶向外显子
‑
外显子连接处相对应;(b)对所述扩增子进行测序以产生多个读段;(c)将所述读段与参考序列进行比对,所述参考序列包含与所述驱动基因的所述所靶向外显子
‑
外显子连接处相对应的所述扩增子的核酸序列;(d)确定与每个所靶向外显子
‑
外显子连接处相对应的每个扩增子的读段数量;(e)将每个扩增子的所述读段数量除以所述驱动基因的所述扩增子之中的最大读段数量以得到每个扩增子的经归一化读段计数;(f)对所述扩增子的所述经归一化读段计数应用基线校正以形成经校正读段计数,其中所述基线校正使用基于多个正常样品的扩增子的读段计数的基线值;(g)确定与所述驱动基因的5'端相对应的扩增子的经校正读段计数和
与所述驱动基因的3'端相对应的扩增子的经校正读段计数之间的不平衡;以及(h)基于所述不平衡来检测所述驱动基因中的基因融合。
7.根据示例性实施例,提供了一种用于检测基因融合的系统,所述系统包括机器可读存储器和与所述存储器连通的处理器,其中所述处理器被配置成执行机器可读指令,所述机器可读指令当由所述处理器执行时使所述系统执行方法,所述方法包括:(a)在所述处理器处接收通过在引物池存在的情况下对核酸样品进行扩增而产生的多个扩增子的多个核酸序列读段,所述引物池包含靶向驱动基因的多个外显子
‑
外显子连接处的引物,其中所述扩增子与所述外显子
‑
外显子连接处相对应;(b)将所述读段与参考序列进行比对,所述参考序列包含与所述驱动基因的所述所靶向外显子
‑
外显子连接处相对应的所述扩增子的核酸序列;(c)确定与每个所靶向外显子
‑
外显子连接处相对应的每个扩增子的读段数量;(d)将每个扩增子的所述读段数量除以所述驱动基因的所述扩增子之中的最大读段数量以得到每个扩增子的经归一化读段计数;(e)对所述扩增子的所述经归一化读段计数应用基线校正以形成经校正读段计数,其中所述基线校正使用基于多个正常样品的扩增子的读段计数的基线值;(f)确定与所述驱动基因的5'端相对应的扩增子的经校正读段计数和与所述驱动基因的3'端相对应的扩增子的经校正读段计数之间的平衡;以及(f)基于所述不平衡来检测所述驱动基因中的基因融合。
8.根据示例性实施例,提供了一种非暂时性机器可读存储介质,所述非暂时性机器可读存储介质包括指令,所述指令当由处理器执行时使所述处理器执行用于检测基因融合的方法,所述方法包括:(a)在所述处理器处接收通过在引物池存在的情况下对核酸样品进行扩增而产生的多个扩增子的多个核酸序列读段,所述引物池包含靶向驱动基因的多个外显子
‑
外显子连接处的引物,其中所述扩增子与所述外显子
‑
外显子连接处相对应;(b)将所述读段与参考序列进行比对,所述参考序列包含与所述驱动基因的所述所靶向外显子
‑
外显子连接处相对应的所述扩增子的核酸序列;(c)确定与每个所靶向外显子
‑
外显子连接处相对应的每个扩增子的读段数量;(d)将每个扩增子的所述读段数量除以所述驱动基因的所述扩增子之中的最大读段数量以得到每个扩增子的经归一化读段计数;(e)对所述扩增子的所述经归一化读段计数应用基线校正以形成经校正读段计数,其中所述基线校正使用基于多个正常样品的扩增子的读段计数的基线值;(f)确定与所述驱动基因的5'端相对应的扩增子的经校正读段计数和与所述驱动基因的3'端相对应的扩增子的经校正读段计数之间的平衡;以及(f)基于所述不平衡来检测所述驱动基因中的基因融合。
9.根据示例性实施例,提供了一种用于检测基因融合的方法,所述方法包括:(a)在引物池存在的情况下对核酸样品进行扩增以产生多个扩增子,所述引物池包含靶向驱动基因的多个外显子
‑
外显子连接处的引物,其中所述扩增子与所述外显子
‑
外显子连接处相对应;(b)对所述扩增子进行测序以产生多个读段;(c)将所述读段与参考序列进行比对;(d)通过将与每个扩增子相对应的读段数量除以所述驱动基因的所述扩增子之中的最大读段数量来对读段数量进行归一化以得到每个扩增子的经归一化读段计数;(e)对所述驱动基因的所述扩增子的所述经归一化读段计数应用基线校正以形成经校正读段计数,其中所述扩增子的所述经校正读段计数通过所述经归一化读段计数除以所述扩增子的基线值的log2确定;(f)计算每个经校正读段计数的二元分割评分以提供与所述多个扩增子相对应的多个二元分割评分;以及(g)基于与最大绝对二元分割评分相对应的扩增子指数来确定
所述基因融合的预测断点。
10.根据示例性实施例,提供了一种用于检测基因融合的系统,所述系统包括机器可读存储器和与所述存储器连通的处理器,其中所述处理器被配置成执行机器可读指令,所述机器可读指令当由所述处理器执行时使所述系统执行方法,所述方法包括:(a)在引物池存在的情况下对核酸样品进行扩增以产生多个扩增子,所述引物池包含靶向驱动基因的多个外显子
‑
外显子连接处的引物,其中所述扩增子与所述外显子
‑
外显子连接处相对应;(b)对所述扩增子进行测序以产生多个读段;(c)将所述读段与参考序列进行比对;(d)通过将与每个扩增子相对应的读段数量除以所述驱动基因的所述扩增子之中的最大读段数量来对读段数量进行归一化以得到每个扩增子的经归一化读段计数;(e)对所述驱动基因的所述扩增子的所述经归一化读段计数应用基线校正以形成经校正读段计数,其中所述扩增子的所述经校正读段计数通过所述经归一化读段计数除以所述扩增子的基线值的log2确定;(f)计算每个经校正读段计数的二元分割评分以提供与所述多个扩增子相对应的多个二元分割评分;以及(g)基于与最大绝对二元分割评分相对应的扩增子指数来确定所述基因融合的预测断点。
11.根据示例性实施例,提供了一种非暂时性机器可读存储介质,所述非暂时性机器可读存储介质包括指令,所述指令当由处理器执行时使所述处理器执行用于检测基因融合的方法,所述方法包括:(a)在引物池存在的情况下对核酸样品进行扩增以产生多个扩增子,所述引物池包含靶向驱动基因的多个外显子
‑
外显子连接处的引物,其中所述扩增子与所述外显子
‑
外显子连接处相对应;(b)对所述扩增子进行测序以产生多个读段;(c)将所述读段与参考序列进行比对;(d)通过将与每个扩增子相对应的读段数量除以所述驱动基因的所述扩增子之中的最大读段数量来对读段数量进行归一化以得到每个扩增子的经归一化读段计数;(e)对所述驱动基因的所述扩增子的所述经归一化读段计数应用基线校正以形成经校正读段计数,其中所述扩增子的所述经校正读段计数通过所述经归一化读段计数除以所述扩增子的基线值的log2确定;(f)计算每个经校正读段计数的二元分割评分以提供与所述多个扩增子相对应的多个二元分割评分;以及(g)基于与最大绝对二元分割评分相对应的扩增子指数来确定所述基因融合的预测断点。
附图说明
12.本发明的新颖特征在所附权利要求书中具体阐述。通过参考阐述说明性实施例和附图的以下详细描述将获得对特征和优点的更好的理解,在所述附图中:
13.图1展示了靶向eml4
‑
alk融合的引物设计和扩增子的实例。
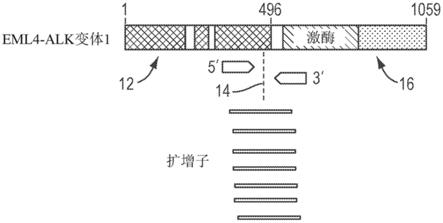
14.图2a展示了融合阴性样品中的近似平衡的3'/5'表达覆盖率模式的实例。
15.图2b展示了融合阳性样品中的显著不平衡的3'/5'表达覆盖率模式的实例。
16.图3展示了用于检测融合的外显子平铺覆盖率分析的实例。
17.图4a展示了正常样品中驱动基因扩增子中的野生型转录物覆盖率模式的实例。
18.图4b示出了融合阳性样品的驱动基因扩增子中的转录物覆盖率模式的实例。
19.图5a是alk基因的所靶向外显子
‑
外显子连接处的经归一化读段计数的绘图的实例。
20.图5b示出了基线校正之后所靶向外显子
‑
外显子连接处的经校正读段计数的实
例,以及正常样品组的经归一化和经基线校正读段计数的绘图。
21.图5c示出了与alk基因的扩增子相对应的二元分割评分z
i
(依据图5b的经基线校正谱计算)的绘图的实例。
22.图6给出了alk基因中每扩增子经归一化读段计数的绘图和每扩增子经基线归一化读段计数的绘图的实例。
23.图7a示出了对照基因itgb7的经基线校正读段计数的绘图的实例。
24.图7b示出了对照基因itgb7的二元分割评分的绘图的实例。
25.图8a示出了对照基因hmbs的经基线校正读段计数的实例。
26.图8b示出了对照基因hmbs的二元分割评分的绘图的实例。
27.图9示出了测试样品的基因alk、itgb7对照基因和hmbs对照基因的二元分割评分的分布的实例。
28.图10示出了来自多个正常样品的用于确定ret基因的基线的经归一化计数的绘图的实例。
29.图11示出了来自多个样品的用于确定braf基因的基线的经归一化计数的绘图的实例。
30.图12示出了在正常样品的背景下两个测试样品的经归一化分子计数和依据其计算的ros1基因的基线的绘图的实例。
31.图13示出了在应用基线校正之前针对两个ros1样品确定的二元分割评分的绘图的实例。
32.图14示出了融合阳性样品(2次重复)142和融合阴性样品(2次重复)144两者的经基线校正分子计数的绘图的实例。
33.图15示出了基线校正之后融合阳性样品重复的二元分割评分的绘图的实例。
34.图16给出了alk基因的融合检测结果的实例。
35.图17给出了ret基因的融合检测结果的实例。
36.图18给出了fgfr3驱动基因的融合检测结果的实例。
37.图19给出了ntrk1基因的融合检测结果的实例。
38.图20给出了已知对于alk的融合为阳性的ffpe样品的外显子平铺融合不平衡和所靶向融合的结果的实例的表格。
39.图21示出了ntrk1基因的经校正读段计数的绘图的实例。
40.图22示出了ret基因的经校正读段计数的绘图的实例。
41.图23示出了ros1基因的经校正读段计数的绘图的实例。
42.图24a示出了alk基因的原始读段计数集和减少的读段集的读段计数的绘图的实例。
43.图24b示出了alk基因的原始读段计数集和减少的读段计数集的经归一化读段计数的绘图的实例。
44.图24c示出了alk基因的原始分子计数集和减少的分子计数集的分子计数的绘图的实例。
45.图24d示出了alk基因的原始分子计数集和减少的分子计数集的经归一化分子计数的绘图的实例。
46.图25是根据实施例的用于基于不平衡分析对融合进行检测的示例性方法的框图。
47.图26是根据实施例的用于生成用于基于不平衡分析对融合进行检测的共有压缩数据的示例性方法的框图。
48.图27是根据实施例的用于流空间共有管线2606的示例性方法的框图。
49.图28示出了可以进行碱基识别的流空间信号测量结果的示例性表示。
50.图29展示了单个家族的流空间信号测量结果的示例性绘图。流指数指示流序列中的第j个流。
51.图30展示了单个家族的共有流空间信号测量的示例性绘图。
52.图31示出了根据各种实施例的核酸测序系统的框图。
具体实施方式
53.根据本技术中体现的教导和原理,提供了用于基于驱动基因中的外显子
‑
外显子连接处的靶向rna测序和5'表达与3'表达之间的不平衡对基因融合进行伴侣未知的检测的新方法、系统和非暂时性机器可读存储介质。
54.在各个实施例中,dna(脱氧核糖核酸)可以被称为由4种类型的核苷酸组成的核苷酸链:a(腺嘌呤)、t(胸腺嘧啶)、c(胞嘧啶)和g(鸟嘌呤),并且所述rna(核糖核酸)包含4种类型的核苷酸:a、u(尿嘧啶)、g和c。某些核苷酸对以互补方式彼此特异性结合(被称为互补碱基配对)。也就是说,腺嘌呤(a)与胸腺嘧啶(t)配对(然而,在rna的情况下,腺嘌呤(a)与尿嘧啶(u)配对),并且胞嘧啶(c)与鸟嘌呤(g)配对。在第一核酸股结合于由与第一股中的核苷酸互补的核苷酸组成的第二核酸股时,两个链结合以形成双股。在各个实施例中,“核酸测序数据”、“核酸测序信息”、“核酸序列”、“基因组序列”、“基因序列”或“片段序列”或“核酸测序读段”或“核酸序列读段”或“序列读段”表示指示核苷酸碱基(例如腺嘌呤、鸟嘌呤、胞嘧啶和胸腺嘧啶/尿嘧啶)在dna或rna的分子(例如全基因组、全转录组、外显子组、寡核苷酸、多核苷酸、片段等)中的次序的任何信息或数据。
55.在各种实施例中,“聚核苷酸”、“核酸”或“寡核苷酸”是指通过核苷间键接合的核苷(包含脱氧核糖核苷、核糖核苷或其类似物)的线形聚合物。通常,聚核苷酸包括至少三个核苷。通常,寡核苷酸的尺寸在数个单体单元(例如3
‑
4个)到数百个单体单元范围内。每当聚核苷酸(例如寡核苷酸)由一连串字母(例如“atgcctg”)表示时,将理解,除非另外指出,否则核苷酸按从左到右的5'
‑
>3'次序并且“a”表示脱氧腺苷,“c”表示脱氧胞苷,“g”表示脱氧鸟苷,且“t”表示胸苷。如在本领域中标准的,字母a、c、g和t可以用于指碱基本身、核苷或包括碱基的核苷酸。
56.如本文中所使用,术语“基因座”是指在染色体或核酸分子上的具体位置。基因座的等位基因位于同源染色体上的相同位点处。
57.如本文中所使用,术语“衔接子”或“衔接子和其补体”以及其派生词通常指任何可以与本公开的核酸分子接合的线形寡核苷酸。任选地,衔接子包含基本上不与样品内至少一个靶序列的3'端或5'端互补的核酸序列。在一些实施例中,衔接子基本上不与样品中任何靶序列的3'端或5'端互补。在一些实施例中,衔接子包含任何基本上不与经扩增的靶序列互补的单链或双链线性寡核苷酸。在一些实施例中,衔接子基本上不与样品的至少一个、一些或全部核酸分子互补。在一些实施例中,合适的衔接子长度在长度为约10
‑
100个核苷
酸、约12
‑
60个核苷酸和约15
‑
50个核苷酸的范围内。衔接子可以包含核苷酸和/或核酸的任何组合。在一些方面中,衔接子可以在一个或多个位置包含一个或多个可裂解基团。在另一方面中,衔接子可以包含与引物(例如通用引物)的至少一部分基本上一致或基本上互补的序列。在一些实施例中,衔接子可以包含条形码或标签以辅助下游编目、鉴别或测序。在一些实施例中,在与经扩增的靶序列接合时,尤其在适合的温度和ph值下在聚合酶和dntp存在下,单股衔接子可以充当用于扩增的底物。
58.如本文中所使用,“dna条形码”或“dna标签序列”和其派生词是指可以充当用于区分或分离样品中多个经扩增的靶序列的
‘
钥匙’的衔接子内的独特短(例如,6
‑
14个核苷酸)核酸序列。出于本公开的目的,dna条形码或dna标签序列可以并入衔接子的核苷酸序列中。
59.在一些实施例中,本公开提供了来自靶核酸分子群的多个靶特异性序列的扩增。在一些实施例中,方法包括将一个或多个靶特异性引物对杂交到靶序列,延伸引物对的第一引物,使来自核酸分子群的延伸第一引物产物变性,将引物对的第二引物杂交到延伸第一引物产物,延伸第二引物以形成双链产物,和远离双链产物消化靶特异性引物对来产生多个经扩增靶序列。在一些实施例中,消化包含从经扩增靶序列中部分消化一种或多种靶特异性引物。在一些实施例中,经扩增靶序列可以连接到一个或多个衔接子。在一些实施例中,衔接子可以包含一个或多个dna条形码或标记序列。在一些实施例中,经扩增靶序列一旦连接到衔接子,可以经历切口平移反应和/或进一步扩增来产生衔接子连接的经扩增靶序列库。
60.在一些实施例中,本公开的方法包含选择性扩增含有多个核酸分子的样品中的靶序列,并将经扩增靶序列连接到至少一个衔接子和/或条形码。用于分子生物学文库制备技术的衔接子和条形码是所属领域的技术人员众所周知的。如本文中所使用的衔接子和条形码的定义与所属领域中使用的术语一致。例如,条形码的使用允许每个多重反应检测和分析多个样品、来源、组织或核酸分子群体。条形码化和经扩增的靶序列含有独特的核酸序列,通常是短的6
‑
15个核苷酸序列,其鉴别和区分一个经扩增的核酸分子与另一个经扩增的核酸分子,即使当减去条形码的两个核酸分子都含有相同的核酸序列时也是如此。衔接子的使用允许以均匀的方式扩增每个经扩增的核酸分子并且有助于减少股偏差。衔接子可以包含通用衔接子或专用衔接子,其都可以在下游使用以执行一个或多个不同的功能。例如,由本文中公开的方法制备的经扩增的靶序列可以接合到可以在下游用作克隆扩增平台的衔接子。衔接子可以充当模板股用于随后使用第二组引物进行扩增,并且因此实现衔接子接合的经扩增的靶序列的通用扩增。在一些实施例中,用于产生扩增子池的靶核酸的选择性扩增可以进一步包括将一个或多个条形码和/或衔接子接合到经扩增的靶序列。合并条形码的能力可以增强样品通量,并且允许同时分析多个样品或材料来源。
61.在本技术案中,“反应限制区域”通常是指可以限制反应的任何区域,并且包含例如“反应室”、“孔”和“微孔”(其中的每一个可以互换使用)。例如,反应限制区域可以包含其中固体基板的物理或化学属性可以允许相关反应的定位的区域,以及可以特异性结合相关分析物的基板表面的离散区域(如具有与这类表面共价连接的寡核苷酸或抗体的离散区域)。反应限制区域可以是中空的或具有明确限定的形状和体积,其可以被制造成基板。这些后面类型的反应限制区域在本文中称为微孔或反应室,并且可以使用任何合适的微制造技术来制造。例如,反应限制区域也可以是没有孔的基板上的基本上平坦的区域。
62.多个限定的空间或反应限制区域可以布置成阵列,并且每个限定的空间或反应限制区域可以与至少一个传感器电连通,以允许检测或测量一个或多个可检测或可测量的参数或特性。此阵列在本文中被称为传感器阵列。传感器可以将反应副产物的存在、浓度或量的变化(或反应物的离子特性的变化)转换成输出信号,所述输出信号可以电子记录,例如以电压水平或电流水平的变化形式,其继而可以被处理以提取关于化学反应或所需关联事件(例如,核苷酸掺入事件)的信息。传感器可以包含至少一个化学敏感性场效应晶体管(“chemfet”),其可以被配置成产生与化学反应的性质或其附近的感兴趣靶标分析物相关的至少一个输出信号。这类特性可以包含反应物、产物或副产物的浓度(或浓度变化),或物理性质的值(或这类值的变化),例如离子浓度。例如,对于限定的空间或反应限制区域的ph值的初始测量或询问可以表示为电信号或电压,其可以被数字化(例如,转换为电信号或电压的数字表示)。这些测量值和表示中的任何一个都可以被认为是原始数据或原始信号。
63.在各种实施例中,短语“碱基空间”是指核苷酸序列的表示。短语“流空间”是指特定核苷酸流的并入事件或非并入事件的表示。例如,流空间可以是表示特定核苷酸流的核苷酸并入事件(如一,“1”)或非并入事件(例如零,“0”)的一系列值。具有非并入事件的核苷酸流可以称为空流,并且具有核苷酸并入事件的核苷酸流可以称为正流。应理解,零和一为非并入事件和核苷酸并入事件的方便表示;但是,任何其它符号或标识可以替代性地用于表示和/或鉴别这些事件和非事件。特定而言,在多个核苷酸在给定位置处并入时,例如对于均聚物拉伸,值可以与核苷酸并入事件数目且因此与均聚物拉伸长度成比例。
64.图1展示了引物对设计和靶向eml4
‑
alk融合的扩增子的实例。对于所靶向融合同种型,引物设计限于已知融合断点处的已知融合产物(即驱动基因与伴侣基因之间的嵌合基因融合)。在此实例中,eml4部分12和alk部分16在断点14处融合。所产生的扩增子跨断点14进行比对,其中每个扩增子的一部分在任一侧上。群体中的常见变异需要多种同种型来支持断点位置的变异。驱动基因可以在许多断点上与数十个基因成为伴侣。
65.图2a和2b展示了检测其中3'/5'表达覆盖率不平衡的融合的实例。在此实例中,alk是驱动基因。在如图2a所示的驱动基因的野生型转录物中,5'的扩增子22和3'的扩增子24在融合阴性样品中具有近似平衡的表达模式。在对于融合为阳性的涉及如图2b所示的驱动基因的样品中,由于融合转录物的表达增强,在驱动基因的5'表达与3'表达26之间将存在显著不平衡,从而有利于后者的表达增加。对于融合阳性样品,仅融合转录物或融合和野生型转录物两者进行表达。
66.图3展示了用于检测融合的外显子平铺覆盖率分析的实例。对于此实例中,驱动基因的每外显子
‑
外显子连接处设计了一个扩增子。扩增子覆盖率模式示出了在外显子6和外显子7的外显子
‑
外显子连接处之后计数增加,这表明存在断点32。
67.图4a和4b示出了跨正常样品(图4a)和融合阳性样品(图4b)中的野生型转录物的基因的覆盖率模式的示意性实例。y轴线表示相对于基线归一化之后的读段计数。x轴线表示与外显子
‑
外显子连接处相对应的内含子指数。图4a示出了跨正常样品的基因的相当均匀的读段覆盖率。图4b示出了在融合阳性样品的断点之后覆盖率急剧增加。
68.在一些实施例中,用于外显子平铺融合检测的扩增子的引物的设计指南包含以下中的一个或多个:
69.·
扩增子长度均匀,优选范围为90
‑
100bp
70.·
gc含量均匀,优选范围为40%
‑
60%
71.·
扩增子放置在每个基因的已知融合断点周围
72.ο至少3个扩增子位于已知断点的每一侧
73.·
不跨越最常见断点的外显子
‑
外显子连接处
74.ο这避免了共享最常见融合的引物(例如:alk exon 20)
75.·
伴侣未知的融合检测的驱动基因优先
76.·
基因的rna表达水平跨几种组织类型
77.·
最小化与其它rna所靶向融合引物的冲突
78.·
在一些实施例中,所述设计针对紧邻基因的非翻译区(utr)发生(例如,在更频繁地在5'端处融合的驱动基因fgfr2和fgfr3(其中融合连接处接近其3putr出现)中发生)的融合断点。在这些情况下,外显子
‑
外显子连接处扩增子可以在utr区处用另外的设计补充,使得保留对断点两侧的表达的足够采样。
79.在一些实施例中,驱动基因的外显子
‑
外显子连接处的扩增子平铺可以与已知断点区域的外显子分开。除了用于扩增子平铺的引物之外,还可以使用被设计成产生已知融合产物的靶向同种型的扩增子的引物。除了使用平铺扩增子检测基因中的其它地方的可能从头断点之外,可以对已知断点的靶向同种型进行测试。在一些实施例中,当对驱动基因的边缘附近的可能断点进行测试时,扩增子平铺可以跨越边缘上的外显子与非翻译区(utr)的边界。
80.在一些实施例中,表1给出了外显子平铺分析的实例。
81.表1.
82.83.在一些实施例中,可以包含更少的基因,如表2的实例中的。
84.表2.
[0085][0086]
在各个实施例中,可以提供基因和多个扩增子的其它组合以供外显子平铺测定。
[0087]
在一些实施例中,给定外显子
‑
外显子连接处的5'引物和3'引物各自具有分子标签。为了标识单独多核苷酸分子,将分子标签分别附加到5'引物和3'引物,包含附加到5'引物的前缀标签和附加到3'引物的后缀标签。单独多核苷酸分子用独特分子标签进行标记,在pcr反应中扩增并测序,从而产生外显子平铺扩增子。给定所靶向融合的外显子平铺扩增子可以包含5'端的前缀标签和3'端的后缀标签。pcr扩增和测序可以产生多个扩增子,从而导致当存在对应外显子
‑
外显子连接处时每原始标记的多核苷酸分子具有多个序列读段。所述独特分子标签用于标识源自同一多核苷酸分子的序列读段,并将其分类为具有相同标签序列的家族。
[0088]
家族或分子家族是指具有相同独特分子标签的一组序列读段。家族大小是家族中的序列读段数量。功能性家族是成员数量大于最小家族大小的家庭。最小家庭大小可以是任何整数值。例如,最小家庭大小可以是三个或更大。与特定扩增子相对应的分子计数是针对所述扩增子计数的家族的数量。
[0089]
在图5a
‑
5c中,针对基因显示的x轴线标记的“内含子”的数量与转录物rna的外显子
‑
外显子连接处的指数相对应(即,内含子指数不代表dna链中的物理内含子序列)。图5a
–
5c中的绘图的圆圈指示引物靶向的所靶向外显子
‑
外显子连接处,使得在这些位置处产生扩增子覆盖率数据。扩增子的位置的数量小于或等于基因中的外显子
‑
外显子连接处的总数。
[0090]
图5a是alk基因中的所靶向外显子
‑
外显子连接处的经归一化读段计数的绘图的实例。在图5a中,激酶区中的内含子指数26处的扩增子示出经归一化读段数量比相邻扩增子的那些低得多。扩增子覆盖率的此下降的可能原因包含受损的扩增子引物结合、导致外显子跳跃或其它技术偏差的剪接同种型。此扩增子的覆盖率在包含正常样品的所有样品中通常将在系统方面较低。基线校正减轻了此问题,如图5b中的经校正读段计数所示。
[0091]
在一些实施例中,应用了不平衡分析以基于从外显子平铺扩增子获得的读段计数对融合进行监测。不平衡分析的输入是与测试样品中的基因的外显子平铺中所靶向外显子
‑
外显子连接处相对应的扩增子的读段数量或覆盖率。例如,输入信息可以在bam文件中
提供。基线是依据从对于基因融合为阴性的许多正常样品中获得的基的因扩增子的读段数量确定的。步骤的次序是示例性的,并且在各个实施例中步骤的不同次序是可能的。
[0092]
1)通过将读段数量除以基因的扩增子之中的最大读段数量对基因的每个扩增子的读段数量进行归一化,以得到每个扩增子的经归一化读段计数。图5a是alk基因的每个所靶向外显子
‑
外显子连接处的经归一化读段计数的绘图的实例。
[0093]
2)由一组40
‑
60个正常样品(即,利用正交方法验证的融合阴性样品)计算基线,以供基线校正使用。每个正常样品中的基因扩增子的读段计数通过将每个基因扩增子的读段数量除以正常样品中覆盖率最高的基因扩增子的读段数量进行归一化。每个扩增子的基线值计算为跨所有正常样品的扩增子的经归一化值的中值。可替代地,可以计算基因的每个扩增子的经归一化读段计数的均值以得到基线值。
[0094]
3)对测试样品基因的扩增子的经归一化读段计数应用基线校正,以形成经校正读段计数。对于测试样品基因的每个扩增子,经校正读段计数可以通过以下计算:
[0095]
经校正读段计数=log2[测试样品中的扩增子的经归一化读段计数
÷
扩增子的基线值]
[0096]
图5b示出了基线校正之后每个所靶向外显子
‑
外显子连接处的校正读段计算52的绘图,以及一组正常样品54的经归一化和经基线校正读段计数的绘图的实例。图5b示出了图5a中示出的alk基因的每个外显子
‑
外显子连接处的几个经校正读段计数52的绘图的实例。
[0097]
4)计算二元分割评分。对于扩增子测量结果x1到x
n
,每个x
i
表示与第i个外显子
‑
外显子连接处相对应的第i个扩增子的经校正读段计数,其中x是经校正读段计数的总数,并且n为一系列扩增子的数量。例如,在图5b中,经校正读段计数的每个数据点与从x1到x
n
的一系列扩增子测量结果相对应,其中n=14。二元分割评分可以如下计算:
[0098]
a.计算从第一扩增子到第i扩增子的扩增子测量结果的部分和s
i
:
[0099]
s
i
=x1+
…
+x
i
[0100]
b.计算从第一扩增子到第n扩增子的所有扩增子测量结果的总和s
n
,其中n是经校正读段计数的总数:
[0101]
s
n
=x1+
…
+x
n
[0102]
c.计算二元分割评分z
i
:(对于测量结果x1…
x
n
‑1)
[0103][0104]
图5c示出了与alk基因的扩增子相对应的二元分割评分z
i
的绘图的实例。
[0105]
d.找到与最大绝对二元分割评分|z
i
|相对应的扩增子指数i。预测断点b是与基因的最大绝对二元分割评分z
max
相对应的扩增子指数i
max
。基因中的物理融合断点可以位于与最大绝对二元分割评分z
max
相对应的扩增子指数i
max
与下一扩增子指数(i
max
+1)之间的范围内。对于图5c的实例,断点被指示为介于内含子指数15与20之间或在与最大绝对二元分割评分z
max
=|z9|相对应的扩增子指数=9之后。
[0106]
5)计算不平衡评分。阵列u被定义为n个扩增子的阵列,其中每扩增子具有经归一
化读段计数,并且阵列v被定义为来自正常样品的扩增子的n个经基线归一化读段计数的阵列,其中n是测量的扩增子的总数。n个经基线归一化读段计数的阵列包含在步骤2)中计算的每个扩增子的基线值。图6给出了每扩增子62的经归一化读段计数的绘图和每扩增子64的经基线归一化读段计数的绘图的实例。圆圈指示测量的扩增子的经归一化读段计数。不平衡评分如下计算:
[0107]
a.基于阵列v中的经基线归一化读段计数(基线值)计算预期不平衡值。在部分和{v[(1+b):n])}中,b为通过测试样品的二元分割评分确定的预测断点,并且经基线归一化读段计数的总和从阵列元素(1+b)到阵列元素n计算。
[0108]
预期不平衡=总和{v[(1+b):n])}/总和[v(1:n)]
[0109]
预期不平衡的此计算用于确定步骤5)c中的场景伴侣基因
–
驱动基因的3'/5'不平衡评分,其中驱动基因位于融合的3'端处。
[0110]
b.基于阵列u中的经归一化读段计数计算观察到的不平衡值。在部分和{u[(1+b):n])}中,b为通过测试样品的二元分割评分确定的预测断点,并且经归一化读段计数的总和从阵列元素(1+b)到阵列元素n计算。
[0111]
观察到的不平衡=总和{u[(1+b):n])}/总和[u(1:n)]
[0112]
观察到的不平衡的此计算用于确定步骤5)c中的场景伴侣基因
–
驱动基因的3'/5'不平衡评分,其中驱动基因位于融合的3'端处。
[0113]
c.不平衡评分是观察到的不平衡值与预期不平衡值之比。
[0114]
不平衡评分=观察到的不平衡/预期不平衡
[0115]
d.在驱动基因可以在5'端处融合的情况下,步骤5)a和5)b的不平衡计算如下反转:
[0116]
预期不平衡=总和{v[1:b])}/总和[v(1:n)]
[0117]
观察到的不平衡=总和{u[1:b])}/总和[u(1:n)]
[0118]
预期不平衡和观察到的不平衡的这些计算用于确定步骤5)c中的场景驱动基因
–
伴侣基因的5'/3'不平衡评分,其中驱动基因位于融合的5'端处。
[0119]
6)应用威尔科克森秩检验(wilcoxon rank test)以将测试样品的二元分割评分与对照基因的二元分割评分进行比较并确定p值。图7b示出了如步骤4)a到4)c中所述计算的对照基因itgb7的二元分割评分的绘图的实例。图7a示出了在itgb7的基线校正之后每个所靶向外显子
‑
外显子连接处的经校正读段计数的绘图的实例。图8b示出了如步骤4)a到4)c中所述计算的对照基因hmbs的二元分割评分的绘图的实例。图8a示出了在hmbs的基线校正之后每个所靶向外显子
‑
外显子连接处的经校正读段计数的绘图的实例。图9示出了测试样品的基因alk、itgb7对照基因和hmbs对照基因的二元分割评分的分布。p值相对于itgb7对照基因为9.3e
‑
06并且相对于hmbs对照基因为4.0e
‑
04。对于两个对照基因,可以使用相应p值中的较大值作为p值,以评价融合检测的不平衡的显著性。在一些实施例中,可以使用单个对照基因以产生单个p值。
[0120]
7)将对不平衡评分和p值应用阈值以检测驱动基因融合。例如,当计算的不平衡评分大于或等于基因的不平衡评分阈值并且计算的p值小于或等于p值阈值时,决策逻辑可以指示阳性不平衡识别。可以使用其它数学上等效的表达式来应用阈值。例如,
‑
log10(p值)可以大于或等于
‑
log10(p值阈值)。不平衡评分阈值和p值阈值可以是基因特异性的,因为
基因在长度、公共断点定位和其产生的表达不平衡模式方面显著不同。不同基因在表达水平方面可能不同,因此可以为特定基因设置阈值。可以使用对来自真值集的已知阳性和阴性样品进行测试来设置特定基因的阈值。表3给出了用于表达不平衡分析阳性识别的可调整基因特异性阈值的实例。
[0121]
表3
[0122][0123]
在一些实施例中,对照样品(例如,itgb7或hmbs)的扩增子的读段计数进行归一化和基线校正,如以上步骤1)到3)所述。
[0124]
二元分割评分的背景信息由olshen、adam b.等人于“用于分析基于阵列的dna拷贝数量数据的圆形二元分割(circular binary segmentation for the analysis of array
‑
based dna copy number data)”,《生物统计学(biostatistics)》(2004),第5卷,第4期,第557
‑
572页中描述。
[0125]
在一些实施例中,与外显子平铺扩增子相对应的序列读段包含分子标签。这些序列读段可以分组到共享公共分子标签的家族中。可以针对与外显子
‑
外显子连接处相对应的扩增子对家族进行计数以形成分子计数。不平衡分析方法步骤1)到7)可以应用于每个外显子平铺扩增子的分子计数或家族计数,而不是读段计数或读段数量。
[0126]
在一些实施例中,基线可以依据通过对来自各种组织,如乳房、肺部、结肠、脑部、皮肤和前列腺的多个正常样品进行测试收集的读段计数数据来确定。例如,用于产生基线的测量结果的样品的数量可以是每个基因基线15个到64个ffpe样品,其中中值为59。可以使用更大数量的正常样品。正常样品的扩增子的读段计数可以针对特定基因进行归一化,如以上步骤2)中所述。将每个样品中的每个基因相对于所述样品中的所述基因的测量的最大读段计数进行归一化。可以计算基因的每个扩增子的经归一化读段计数的中值以得到基线。可替代地,可以计算基因的每个扩增子的经归一化读段计数的均值以得到基线。如果所有扩增子的读段计数覆盖率充足,则在基因的基线计算中包含正常样品。例如,每扩增子的经中值归一化覆盖率小于0.1的正常样品可以从基线中排除。图10
‑
11给出了来自多个正常样品的用于确定基线的经归一化计数的实例。还示出了中值和均值。图10给出了来自多个正常样品的用于确定ret基因的基线的经归一化计数的实例。图11给出了来自多个样品的用于确定braf基因的基线的多个样品的经归一化计数的实例。
[0127]
基线校正可以校正数据中的系统变异,如gc偏差、扩增子特异性变异等。图12和13示出了基线校正之前ros1基因的融合阳性样品的结果的实例。图12示出了阳性测试样品122和基线124的两个重复的经归一化分子计数。图13示出了在应用基线校正之前针对两个
样品确定的二元分割评分。图13示出了与预测断点相对应的扩增子指数23(即,从基因5'开始的第23个外显子
‑
外显子连接处)处的最大绝对二元分割评分。图14和15示出了基线校正之后ros1基因的融合阳性样品的两次重复和融合阴性样品的两次重复的结果的实例。图14示出了融合阳性样品(2次重复)142和融合阴性样品(2次重复)144两者的经基线校正分子计数log2(测试样品中的经归一化分子计数/扩增子的基线值)的绘图。图15示出了基线校正之后融合阳性样品重复的二元分割评分。图15示出了与预测断点相对应的扩增子指数21处的最大绝对二元分割评分。预测断点已由于基线校正而偏移,从而产生对断点的更准确的预测。
[0128]
图16
‑
19给出了不同驱动基因的融合检测结果的实例。这些结果是依据对来自特定基因的真值集的已知阳性和阴性样品产生的。用各个背景的正常样品(融合阴性肺部样品)将阳性样品向下稀释到2%
‑
20%的浓度,从而例示测定的检测极限(lod)范围。图16
‑
19示出了表示单个测试样品的点的绘图,其中x轴线为不平衡评分并且y轴线为p值的负log10。表示每基因的指定检测阈值的虚竖线和水平线将点分成具有右上正识别象限的象限(即,不平衡评分>不平衡阈值并且p值<p值阈值)。加号符号表示已知融合阳性样品,并且圆点表示已知融合阴性ffpe样品。右上角象限中的样品被称为融合阳性。图16给出了alk基因的结果的实例。这些结果示出了对实体瘤ffpe样品(福尔马林固定的石蜡包埋的)中的alk基因的阳性(稀释到2%的6个alk融合细胞系样品和稀释到20%的2个alk融合ffpe临床研究样品)和阴性进行的良好分类。类似地,图17给出了ret基因的结果的实例。这些结果示出了对ret基因的融合阳性(稀释到2%的5个ret融合细胞系样品和稀释到20%的1个ret ffpe临床研究样品)和阴性样品进行的总体良好分类。图18给出了在5'端处与3'端处的伴侣基因更频繁地融合的fgfr3驱动基因的结果的实例。在此基因中,沿x轴线的不平衡评分表示反向5'/3'不平衡测量,如以上关于步骤5)d所描述的。所述绘图示出了对阳性(稀释到20%的6个fgfr3
‑
tacc3细胞系样品)和阴性ffpe样品的良好分类。结果示出与其它基因相比fgfr3阳性值的不平衡值较低,因此可以应用较低阈值以在维持测定的高灵敏度的同时对阳性进行正确分类。图19给出了ntrk1基因的结果的实例。对四个ntrk1融合细胞系样品进行了测试。结果示出阳性样品正确分类。
[0129]
在一些实施例中,可以应用各种阈值以基于不平衡分析来识别基因融合。根据步骤7),可以应用p值和不平衡评分的基因特异性阈值。例如,p值的基因特异性阈值可以在0.05到0.1的范围内,并且不平衡评分的基因特异性阈值可以在1.5到3.5的范围内。在一些实施例中,可以应用另外的阈值,包含每扩增子的平均读段计数≥30、每扩增子的平均分子计数≥3并且侧接预测断点的扩增子的数量≥2。
[0130]
在一些实施例中,当所述信息可用时,不平衡识别的结果可以与所靶向同种型识别的结果组合。表4中给出了将不平衡识别与靶向同种型识别组合以得到报告的融合识别的实例。
[0131]
表4
[0132]
靶向同种型识别不平衡识别融合识别
‑
报告阳性负阳性阳性阳性阳性负负负
负阳性阳性
[0133]
图20给出了已知对于alk的融合为阳性的ffpe样品的外显子平铺融合不平衡和所靶向融合的结果的实例的表格。第二列中的术语“外显子15
‑
20”指示与最大绝对二元分割评分z
max
相对应的断点的外显子位置的预测范围。针对所靶向融合同种型检测到的断点的外显子位置由第三列中的框2002中的数字表示。第二列中的断点的预测范围与列3的框2002中的所靶向融合检测到的断点的外显子位置一致。与所靶向融合检测相比,所有样品数量的外显子平铺融合不平衡结果预测了外显子
‑
外显子融合的正确范围。
[0134]
图21示出了ntrk1基因的经校正读段计数的绘图的实例。ntrk1清楚地示出了激酶结构域附近的4个扩增子以预期水平表达并且5'端附近的3个另外的扩增子明显表达不足的不平衡特征。在一些实施例中,覆盖另外的外显子
‑
外显子连接处的更多扩增子可以减少假阴性并改进融合不平衡检测。在一些实施例中,调整不平衡评分和/或p值的基因特异性阈值可以优化融合不平衡检测。
[0135]
图22示出了ret基因的经校正读段计数的绘图的实例。不平衡评分为1.906,p值为0.0027。图23示出了ros1基因的经校正读段计数的绘图的实例。不平衡评分为3.399,并且p值为6e
‑
04。这两个实例都满足对融合不平衡检测的阈值要求。
[0136]
在一些实施例中,bam文件中的读段计数或分子计数可被子采样为原始计数的分数。例如,分数的范围可以为原始计数的2%到20%。子采样可以随机选择读段。经子采样的扩增子可以被重新映射和计数以形成减少的外显子平铺读段计数集或减少的外显子平铺分子计数集。然后可以对减少的外显子平铺读段计数集或减少的外显子平铺分子计数集应用步骤1)到7)中描述的不平衡分析。图24a
‑
24d示出了alk基因的原始读段计数集和减少的读段计数集的实例。图24a和24b中的绘图给出了读段计数,并且图24c和24d中的绘图给出了分子计数。这些绘图示出当使用减少的读段集时覆盖率谱得以保留。表5示出了减少的读段集的外显子平铺融合不平衡的结果的实例。ppv为阳性预测值,fp为假阳性,fn为假阴性,并且tp为真阳性。
[0137]
表5.
[0138]
[0139]
这些结果示出,减少的读段集可以在融合不平衡检测中提供可比的性能。子采样还提供了读段数据的压缩,如表5的“总经映射读段”列所示。减少的读段集需要更少存储器以进行存储。此外,减少的读段集需要更少的计算以进行不平衡分析,因为存在更少的读段来进行分析。对于所述方法在计算机上的实施,存储器的节省和计算负载的降低提高了计算性能。
[0140]
图25是根据实施例的用于基于不平衡分析对融合进行检测的示例性方法的框图。可以通过核酸测序装置向处理器提供流空间信号测量结果。在一些实施例中,每个流空间信号测量值表示响应于在传感器阵列的微孔中样品核酸的流动的核苷酸的并入或非并入测量的信号的幅度或强度。对于并入事件,信号幅度取决于在一个流中并入的碱基数目。对于均聚物,信号幅度随着均聚物长度的增加而增加。处理器可以应用碱基识别器2502以通过对流空间信号测量结果进行分析来产生序列读段的碱基识别。
[0141]
图28示出了可以进行碱基识别的流空间信号测量结果的示例性表示。在此实例中,x轴线示出了流指数和在流序列中流动的核苷酸。图中的条形示出来自传感器阵列中微孔的特定位置的每个流的流空间信号测量结果的幅度。流空间信号测量结果可以是原始采集数据或已经处理的数据,例如通过缩放、背景过滤、归一化、信号衰减校正和/或相位误差或效果的校正等。碱基识别可以通过分析任何合适的信号特征(例如,信号幅度或强度)来进行。与本发明的教示一起使用的传感器阵列、信号处理和碱基识别的结构和/或设计可以包含2013年4月11日公开的美国专利申请公开案第2013/0090860号中描述的一个或多个特征,其以全文引用的方式并入本文中。
[0142]
一旦序列读段的碱基序列确定,就可以向映射器2504提供序列读段,例如以为未经映射的bam文件的形式。在一些实施例中,映射器2504将序列读段与包含所靶向外显子
‑
外显子连接处的参考序列和对照基因参考序列进行比对以确定比对的序列读段和相关联的映射质量参数。参考序列和对照基因参考序列可以使用fasta文件格式或另一种合适的文件格式以文件形式提供。与本发明的教示一起使用的用于比对序列读段的方法可以包含2012年8月2日公开的美国专利申请公开案第2012/0197623号中描述的一个或多个特征,该公开以全文引用的方式并入本文中。
[0143]
在一些实施例中,比对的序列读段可以提供给不平衡分析管线2512。不平衡分析管线2512可以应用以上所述的步骤1)到7)。可以将应用于不平衡评分和p值以检测驱动基因融合的阈值的结果提供给融合识别器2510。融合识别器2510可以基于应用于不平衡分析的阈值的结果提供融合识别。在一些实施例中,当所述信息可用时,不平衡识别的结果可以与所靶向同种型识别的结果组合。以上表4中给出了将不平衡识别与所靶向同种型识别组合以得到报告的融合识别的实例。用于检测用于与本教导一起使用的所靶向融合的方法可以包含于2018年9月20日提交的美国专利申请第16/136,463号中描述的一个或多个特征,所述美国专利申请通过引用以其整体并入本文。
[0144]
在一些实施例中,与外显子平铺扩增子相对应的序列读段包含分子标签。这些序列读段可以分组到共享公共分子标签的家族中。可以针对与外显子
‑
外显子连接处相对应的扩增子对家族进行计数以形成分子计数。图26是根据实施例的用于生成用于基于不平衡分析对融合进行检测的共有压缩数据的示例性方法的框图。可以将经比对的序列读段提供给流空间共有管线2606,例如,以经映射的bam文件的形式。可以将经映射的共有bam文件提
供给第二级压缩器2608。可以将家族计数或分子计数提供给不平衡分析管线2512。不平衡分析管线2512可以对分子计数或家族计数应用不平衡分析方法步骤1)到7),以用于外显子平铺扩增子而不是读段计数或读段数量。
[0145]
bam文件格式结构在2014年9月12日的被称为“bam规范(bam specification)”的“序列比对/映射格式规范(sequence alignment/map format specification)”(https://github.com/samtools/hts
‑
specs)中进行了描述。如本文中所描述,“bam文件”是指与bam格式兼容的文件。如本文中所述,“未经比对的”bam文件是指不含有经比对的序列读段信息或映射质量参数的bam文件,并且“经映射的”bam文件是指含有经比对的序列读段信息和映射质量参数的bam文件。如本文所述,“共有”bam文件是指含有共有压缩数据的bam文件。
[0146]
在一些实施例中,具有分子标记的序列读段的读段结构可以包含从5'端开始的文库密钥、条形码序列、条形码衔接子、前缀分子标签、序列模板、后缀分子标签、和p1衔接子。碱基识别可以包含从序列读段的其余部分修剪文库密钥、条形码序列和条形码衔接子,并将其存储在bam文件格式的读段组标头@rg的密钥序列(ks)标签字段中。碱基识别可以包含从序列读段修剪p1衔接子并将其存储在bam标头的注释行@co中。
[0147]
在一些实施例中,碱基识别器2502可以被配置成检测标签结构并从序列读段修剪标签。经修剪的标签可以存储在自定义标签zt(例如用于前缀标签)和yt(例如用于后缀标签)的字段中的bam读段组标头(@rg)中。由于读段组标头与模板的序列读段数据相关联,因此可以维持标签与家族组的关联的完整性。可以在没有前缀标签或后缀标签的情况下对模板序列应用与参考序列的后续映射或比对。这降低了将标签的一部分错误映射到参考序列的可能性。
[0148]
在一些实施例中,标签序列可以包含随机碱基的子集和已知碱基的子集。标签修剪方法可能需要序列读段的标签部分中的碱基序列与已知碱基匹配。标签修剪方法可以选择碱基的数量与标签的已知长度相等的碱基串。在一些实施例中,标签修整方法可以检测并校正标签中的测序误差,如插入和缺失。标签中的校正测序误差可以提供更准确的家族标识。
[0149]
在一些实施例中,经映射的bam文件可以存储多个序列读段、流空间信号测量结果的多个向量和与序列读段相对应的多个序列比对。经映射的bam文件可以将流空间信号测量结果的向量存储在自定义标签字段zm中。经映射的bam文件可以将模型参数存储在自定义标签字段zp中。经映射的bam文件可以将与序列读段相关联的分子标签序列存储在bam读段组标头中,如上所述。经映射的bam文件可以存储在存储器中并提供给流空间共有管线2606。在一些实施例中,可以使用其它文件格式来存储多个序列读段、流空间信号测量结果的多个向量、多个序列比对和与序列读段相对应的分子标签序列。
[0150]
图27是根据实施例的用于流空间共有管线2606的示例性方法的框图。分组操作302可以使用分子标签序列信息来标识序列读段的家族和对应流空间信号测量结果。分组操作302可以对与序列读段相关联的分子标签序列进行比较并且应用分组阈值。例如,分组阈值的标准可能需要一组序列读段的成员的所有标签序列的标签序列同一性为100%。确定为共享公共标签序列的序列读段和对应流空间信号测量结果通过满足分组阈值的标准而被分组到给定家族中,其中所述公共标签序列对所述家族是唯一的。每个家族的成员的数量将是分组在家族中的序列读段的数量。在一些实施例中,不具有至少最少数量的成员
的家族将不会进行进一步处理并且可以从存储器中移除。用于基于用于与本教导一起使用的分子标签序列对序列读段进行分组的方法可以包含于2016年12月15日发表的美国专利申请公开第2016/0362748号中描述的一个或多个特征,所述美国专利申请公开通过引用以其整体并入本文。
[0151]
在一些实施例中,流空间共有压缩器304可以基于每个分组的家族的流空间信号测量结果如下确定共有压缩数据:
[0152]
a.计算每个分组的家族的流空间信号测量结果的向量的算数平均值,以形成每个家族的共有流空间信号测量结果的向量。
[0153]
b.计算每个家族的流空间信号测量结果的向量的标准偏差,以形成每个家族的标准偏差的向量。
[0154]
在一些实施例中,流空间共有压缩器304可以接收与流空间信号测量结果的每个向量相对应的至少一个模型参数。流空间共有压缩器304可以计算所述家族的模型参数的算数平均值以形成所述家族的至少一个共有模型参数。模型参数可以用于碱基识别,如下所述。在一些实施例中,模型参数可以包含流空间信号测量结果的每个向量的不完整扩展(incomplete extension,ie)参数和结转(carry forward,cf)参数。流空间共有压缩器304可以计算每个家族的ie参数的算数平均值和cf参数的算数平均值,以形成每个家族的共有ie参数和共有cf参数。
[0155]
在一些实施例中,碱基识别器2502可应用于每个家族的共有流空间信号测量结果的向量以生成相应家族的共有碱基序列。共有碱基序列在本文中也被称为共有序列读段。共有模型参数可以用于应用用于碱基识别的模型。例如,每个家族的共有不完全扩展(ie)参数和共有结转(cf)参数可以提供给碱基识别器2502。碱基识别可以包含于2013年4月11日发表的美国专利申请公开第2013/0090860号和/或于2012年5月3日发表的美国专利申请公开第2012/0109598号中描述的一个或多个特征,所述美国专利申请公开通过引用以其整体并入本文。共有碱基序列的共有序列比对可以通过将共有碱基序列与家族中映射质量最高的序列读段进行比较来确定。如果共有碱基序列与映射质量最高的序列读段匹配,则选择对应序列比对作为共有序列比对。如果共有碱基序列与家族中映射质量最高的序列读段不匹配,则映射器2504可以将共有碱基序列与所靶向融合参考序列和对照基因参考序列进行比对以确定共有序列比对。用于对共有序列读段进行比对的方法可以包含于2012年8月2日公布的美国专利申请公开第2012/0197623号中描述的一种或多种特征,该公开以全文引用的方式并入本文中。在一些实施例中,平均共有序列读段的约1%可能需要由映射器2504进行的重新布置。
[0156]
在一些实施例中,处理器可以将每个家族的共有压缩数据以压缩数据结构的形式存储在存储器中。共有压缩数据可以包含共有序列读段、共有序列比对、共有流空间信号测量结果的向量、标准偏差的向量和每个家族的成员的数量。共有压缩数据可以进一步包含每个家族的共有模型参数集。如果家族已被分为亚家族,则共有压缩数据可以进一步包含共有序列读段、共有序列比对、共有流空间信号测量结果的向量、标准偏差的向量和每个亚家族的成员的数量。在一些实施例中,压缩数据结构可与bam文件格式兼容,以产生经映射共有bam文件。bam规范允许用户定义自定义标签字段。例如,自定义标签字段可以针对用于存储如表6所述的共有压缩数据中的一些的bam文件来定义。
[0157]
表6.
[0158]
bam自定义标签字段数据zm共有流空间信号测量结果zp共有模型参数zs流空间信号测量结果的标准偏差zr家族或亚家族中的序列读段或成员的数量
[0159]
每个家族的原始序列读段、流空间信号测量结果的原始向量和原始模型参数不包含在共有压缩数据中并且可以会从存储器移除。在一些实施例中,压缩数据结构可以使用与bam文件格式不同的格式协议,包含自定义文件格式。
[0160]
图29展示了单个家族的流空间信号测量结果的示例性绘图。流指数指示流序列中的第j个流。经归一化幅度指示流空间信号测量结果的值。绘图符号的类型与特定流下的核苷酸相对应。流空间信号测量结果的此绘图和与共有分子标签相关联的序列读段的单个家族相对应。每种流下的流空间信号测量结果的值聚类在相似值附近。流指数与流空间信号测量结果的向量中的元素指数相对应。此绘图中表示的流空间信号测量结果可以输入到流空间共有压缩器304中。
[0161]
图30展示了单个家族的共有流空间信号测量的示例性绘图。此绘图示出了由对图29中示出流空间信号测量结果进行的共有计算产生的共有流空间信号测量值。绘图符号指示作为家族的共有流空间测量结果的向量的元素的算术平均值。条形指示作为家族的标准偏差的向量的元素的标准偏差。
[0162]
对于双向测序,第一家族可以被指定为用于正向序列读段,并且第二家族可以被指定为用于反向序列读段。正向读段的前缀和后缀标签可以是反向读段的前缀和后缀标签的反向补体,如表7的实例所示。
[0163]
表7
[0164]
读段方向前缀标签后缀标签标签序列读段的数量正向读段actggtactggt10反向读段accagtaccagt10反向读段标签的反向补体ggtactactggt20
[0165]
在一些实施例中,家族可以分裂为亚家族,从而导致每家族有多于一个共有序列读段具有相同的分子标签。可以形成亚家族以供流同步,使得每个亚家族具有用于确定共有流空间测量结果的向量的同步的流空间信号测量结果。当家族内的序列读段中存在变化时,家族可能会分裂为亚家族,使得为每个亚家族生成了共有序列读段。用于与本教导一起使用的分子标记的核酸序列数据的流空间共有压缩的方法可以包含于2018年5月15日提交的美国专利申请第15/979,804号中描述的一个或多个特征,所述美国专利申请通过引用以其整体并入本文。
[0166]
返回到图26,在一些实施例中,可以在融合分析之前对共有压缩数据应用第二级压缩器2608。第二级压缩器2608可以将具有相同分子标签的亚家族组合为包含一个共有序列读段的单一家族。在双向测序读段的一些实施例中,第二级压缩器2608可以如下组合正向序列读段和反向序列读段的家族:
[0167]
1.确定反向补体,反向读段的前缀和后缀标签,以形成反向补体标签,
[0168]
2.将反向补体标签与正向读段标签相匹配,
[0169]
3.将具有匹配标签的正向读段家族和反向读段家族组合为包含一个共有序列读段的一个家族。
[0170]
参照表7,经组合的家族所表示的读段的数量为正向和反向读段家族中的序列读段的数量的总和。可以修改经映射的共有bam文件以包含经组合的家族信息并移除亚家族信息。可以在经映射的共有bam文件的zr字段中输入总和值。第二级压缩器2608为经组合的家族提供了单一共有序列读段。通过消除每个经组合的家族的一个亚家族的共有序列读段,第二级压缩器2608提供了另外的数据压缩。在二级压缩之后,可以将共有压缩数据提供给不平衡分析管线2512。
[0171]
在一些实施例中,本文所述的方法可以至少部分地使用分布式、集群化、远程或云计算资源来执行或实施。压缩序列读段数据以提供共有压缩数据为将数据传输到分布式、集群化、远程或云计算资源中的处理器提供了优势。由于数据的体积减少,因此减少了跨计算资源之间的数据传输接口传输所需的带宽和/或时间。例如,经映射的共有bam文件可以从本地计算资源转移到云计算资源以供融合检测操作。经映射的共有bam文件的大小将显著小于原始经映射的bam文件的大小。经映射的共有bam文件的较小大小将减少跨数据传输接口传输到云计算资源所需的带宽和/或时间。
[0172]
根据示例性实施例,提供了一种用于检测基因融合的方法,所述方法包括:(a)在引物池存在的情况下对核酸样品进行扩增以产生多个扩增子,所述引物池包含靶向驱动基因的多个外显子
‑
外显子连接处的引物,其中所述扩增子与所靶向外显子
‑
外显子连接处相对应;(b)对所述扩增子进行测序以产生多个读段;(c)将所述读段与参考序列进行比对,所述参考序列包含与所述驱动基因的所述所靶向外显子
‑
外显子连接处相对应的所述扩增子的核酸序列;(d)确定与每个所靶向外显子
‑
外显子连接处相对应的每个扩增子的读段数量;(e)将每个扩增子的所述读段数量除以所述驱动基因的所述扩增子之中的最大读段数量以得到每个扩增子的经归一化读段计数;(f)对所述扩增子的所述经归一化读段计数应用基线校正以形成经校正读段计数,其中所述基线校正使用基于多个正常样品的扩增子的读段计数的基线值;(g)确定与所述驱动基因的5'端相对应的扩增子的经校正读段计数和与所述驱动基因的3'端相对应的扩增子的经校正读段计数之间的不平衡;以及(h)基于所述不平衡来检测所述驱动基因中的基因融合。所述确定不平衡的步骤可以进一步包含:计算从第一扩增子到第i扩增子的经校正读段计数x的部分和s
i
,其中s
i
=x1+
…
+x;以及计算从所述第一扩增子到第n扩增子的经校正读段计数的总和s
n
,其中s
n
=x1+
…
+x
n
,其中n是经校正读段计数的总数。所述确定不平衡的步骤可以进一步包含通过以下确定第i扩增子的二元分割评分z
i
:
[0173][0174]
所述确定不平衡的步骤可以进一步包含基于与最大绝对二元分割评分相对应的扩增子指数来确定所述基因融合的预测断点。所述确定不平衡的步骤可以进一步包含基于观察到的不平衡值和预期不平衡值之比来确定不平衡评分。所述预期不平衡值可以基于基
线值的第一阵列,并且所述观察到的不平衡值可以基于经归一化读段计数的第二阵列,其中每个阵列中的阵列元素的数量为n。所述确定不平衡评分的步骤可以进一步包含:(a)计算所述第一阵列的从阵列元素(1+b)到阵列元素n的基线值的第一总和,其中b为预测断点;(b)计算所述第一阵列的从阵列元素1到所述阵列元素n的基线值的第二总和;以及(c)将所述第一总和除以所述第二总和以形成所述预期不平衡值。所述确定不平衡评分的步骤可以进一步包含:(a)计算所述第二阵列的从阵列元素(1+b)到阵列元素n的经归一化读段计数的第一总和,其中b为预测断点;(b)计算所述第二阵列的从阵列元素1到所述阵列元素n的经归一化读段计数的第二总和;以及(c)将所述第一总和除以所述第二总和以形成所述观察到的不平衡值。对于位于所述5'端处的所述驱动基因,所述确定不平衡评分的步骤可以进一步包含:(a)计算所述第一阵列的从阵列元素1到阵列元素b的基线值的第一总和,其中b为预测断点;(b)计算所述第一阵列的从阵列元素1到所述阵列元素n的基线值的第二总和;以及(c)将所述第一总和除以所述第二总和以形成所述预期不平衡值。对于位于所述5'端处的所述驱动基因,所述确定不平衡评分的步骤可以进一步包含:(a)计算所述第二阵列的从阵列元素1到阵列元素b的经归一化读段计数的第一总和,其中b为预测断点;(b)计算所述第二阵列的从阵列元素1到所述阵列元素n的经归一化读段计数的第二总和;以及(c)将所述第一总和除以所述第二总和以形成所述观察到的不平衡值。每个扩增子的所述基线值是可以针对所述多个正常样品的所述扩增子确定的多个经归一化读段计数的中值。所述检测所述基因融合的步骤可以进一步包含通过应用威尔科克森秩检验将多个所述二元分割评分和与对照基因的第二多个扩增子相对应的第二多个二元分割评分进行比较来确定p值。所述检测所述基因融合的步骤可以进一步包含对所述p值应用阈值。所述检测所述基因融合的步骤可以进一步包含对所述不平衡评分应用阈值。
[0175]
根据示例性实施例,提供了一种用于检测基因融合的系统,所述系统包括机器可读存储器和与所述存储器连通的处理器,其中所述处理器被配置成执行机器可读指令,所述机器可读指令当由所述处理器执行时使所述系统执行方法,所述方法包括:(a)在所述处理器处接收通过在引物池存在的情况下对核酸样品进行扩增而产生的多个扩增子的多个核酸序列读段,所述引物池包含靶向驱动基因的多个外显子
‑
外显子连接处的引物,其中所述扩增子与所述外显子
‑
外显子连接处相对应;(b)将所述读段与参考序列进行比对,所述参考序列包含与所述驱动基因的所述所靶向外显子
‑
外显子连接处相对应的所述扩增子的核酸序列;(c)确定与每个所靶向外显子
‑
外显子连接处相对应的每个扩增子的读段数量;(d)将每个扩增子的所述读段数量除以所述驱动基因的所述扩增子之中的最大读段数量以得到每个扩增子的经归一化读段计数;(e)对所述扩增子的所述经归一化读段计数应用基线校正以形成经校正读段计数,其中所述基线校正使用基于多个正常样品的扩增子的读段计数的基线值;(f)确定与所述驱动基因的5'端相对应的扩增子的经校正读段计数和与所述驱动基因的3'端相对应的扩增子的经校正读段计数之间的平衡;以及(f)基于所述不平衡来检测所述驱动基因中的基因融合。所述确定不平衡的步骤可以进一步包含:计算从第一扩增子到第i扩增子的经校正读段计数x的部分和s
i
,其中s
i
=x1+
…
+x
i
;以及计算从所述第一扩增子到第n扩增子的经校正读段计数的总和s
n
,其中s
n
=x1+
…
+x
n
,其中n是经校正读段计数的总数。所述确定不平衡的步骤可以进一步包含通过以下确定第i扩增子的二元分割评分z
i
:
[0176][0177]
所述确定不平衡的步骤可以进一步包含基于与最大绝对二元分割评分相对应的扩增子指数来确定所述基因融合的预测断点。所述确定不平衡的步骤可以进一步包含基于观察到的不平衡值和预期不平衡值之比来确定不平衡评分。所述预期不平衡值可以基于基线值的第一阵列,并且所述观察到的不平衡值可以基于经归一化读段计数的第二阵列,其中每个阵列中的阵列元素的数量为n。所述确定不平衡评分的步骤可以进一步包含:(a)计算所述第一阵列的从阵列元素(1+b)到阵列元素n的基线值的第一总和,其中b为预测断点;(b)计算所述第一阵列的从阵列元素1到所述阵列元素n的基线值的第二总和;以及(c)将所述第一总和除以所述第二总和以形成所述预期不平衡值。所述确定不平衡评分的步骤可以进一步包含:(a)计算所述第二阵列的从阵列元素(1+b)到阵列元素n的经归一化读段计数的第一总和,其中b为预测断点;(b)计算所述第二阵列的从阵列元素1到所述阵列元素n的经归一化读段计数的第二总和;以及(c)将所述第一总和除以所述第二总和以形成所述观察到的不平衡值。对于位于所述5'端处的所述驱动基因,所述确定不平衡评分的步骤可以进一步包含:(a)计算所述第一阵列的从阵列元素1到阵列元素b的基线值的第一总和,其中b为预测断点;(b)计算所述第一阵列的从阵列元素1到所述阵列元素n的基线值的第二总和;以及(c)将所述第一总和除以所述第二总和以形成所述预期不平衡值。对于位于所述5'端处的所述驱动基因,所述确定不平衡评分的步骤可以进一步包含:(a)计算所述第二阵列的从阵列元素1到阵列元素b的经归一化读段计数的第一总和,其中b为预测断点;(b)计算所述第二阵列的从阵列元素1到所述阵列元素n的经归一化读段计数的第二总和;以及(c)将所述第一总和除以所述第二总和以形成所述观察到的不平衡值。每个扩增子的所述基线值是可以针对所述多个正常样品的所述扩增子确定的多个经归一化读段计数的中值。所述检测所述基因融合的步骤可以进一步包含通过应用威尔科克森秩检验将多个所述二元分割评分和与对照基因的第二多个扩增子相对应的第二多个二元分割评分进行比较来确定p值。所述检测所述基因融合的步骤可以进一步包含对所述p值应用阈值。所述检测所述基因融合的步骤可以进一步包含对所述不平衡评分应用阈值。
[0178]
根据示例性实施例,提供了一种非暂时性机器可读存储介质,所述非暂时性机器可读存储介质包括指令,所述指令当由处理器执行时使所述处理器执行用于检测基因融合的方法,所述方法包括:(a)在所述处理器处接收通过在引物池存在的情况下对核酸样品进行扩增而产生的多个扩增子的多个核酸序列读段,所述引物池包含靶向驱动基因的多个外显子
‑
外显子连接处的引物,其中所述扩增子与所述外显子
‑
外显子连接处相对应;(b)将所述读段与参考序列进行比对,所述参考序列包含与所述驱动基因的所述所靶向外显子
‑
外显子连接处相对应的所述扩增子的核酸序列;(c)确定与每个所靶向外显子
‑
外显子连接处相对应的每个扩增子的读段数量;(d)将每个扩增子的所述读段数量除以所述驱动基因的所述扩增子之中的最大读段数量以得到每个扩增子的经归一化读段计数;(e)对所述扩增子的所述经归一化读段计数应用基线校正以形成经校正读段计数,其中所述基线校正使用基于多个正常样品的扩增子的读段计数的基线值;(f)确定与所述驱动基因的5'端相对应
的扩增子的经校正读段计数和与所述驱动基因的3'端相对应的扩增子的经校正读段计数之间的平衡;以及(f)基于所述不平衡来检测所述驱动基因中的基因融合。所述确定不平衡的步骤可以进一步包含:计算从第一扩增子到第i扩增子的经校正读段计数x的部分和s
i
,其中s
i
=x1+
…
+x
i
;以及计算从所述第一扩增子到第n扩增子的经校正读段计数的总和s
n
,其中s
n
=x1+
…
+x
n
,其中n是经校正读段计数的总数。所述确定不平衡的步骤可以进一步包含通过以下确定第i扩增子的二元分割评分z
i
:
[0179][0180]
所述确定不平衡的步骤可以进一步包含基于与最大绝对二元分割评分相对应的扩增子指数来确定所述基因融合的预测断点。所述确定不平衡的步骤可以进一步包含基于观察到的不平衡值和预期不平衡值之比来确定不平衡评分。所述预期不平衡值可以基于基线值的第一阵列,并且所述观察到的不平衡值可以基于经归一化读段计数的第二阵列,其中每个阵列中的阵列元素的数量为n。所述确定不平衡评分的步骤可以进一步包含:(a)计算所述第一阵列的从阵列元素(1+b)到阵列元素n的基线值的第一总和,其中b为预测断点;(b)计算所述第一阵列的从阵列元素1到所述阵列元素n的基线值的第二总和;以及(c)将所述第一总和除以所述第二总和以形成所述预期不平衡值。所述确定不平衡评分的步骤可以进一步包含:(a)计算所述第二阵列的从阵列元素(1+b)到阵列元素n的经归一化读段计数的第一总和,其中b为预测断点;(b)计算所述第二阵列的从阵列元素1到所述阵列元素n的经归一化读段计数的第二总和;以及(c)将所述第一总和除以所述第二总和以形成所述观察到的不平衡值。对于位于所述5'端处的所述驱动基因,所述确定不平衡评分的步骤可以进一步包含:(a)计算所述第一阵列的从阵列元素1到阵列元素b的基线值的第一总和,其中b为预测断点;(b)计算所述第一阵列的从阵列元素1到所述阵列元素n的基线值的第二总和;以及(c)将所述第一总和除以所述第二总和以形成所述预期不平衡值。对于位于所述5'端处的所述驱动基因,所述确定不平衡评分的步骤可以进一步包含:(a)计算所述第二阵列的从阵列元素1到阵列元素b的经归一化读段计数的第一总和,其中b为预测断点;(b)计算所述第二阵列的从阵列元素1到所述阵列元素n的经归一化读段计数的第二总和;以及(c)将所述第一总和除以所述第二总和以形成所述观察到的不平衡值。每个扩增子的所述基线值是可以针对所述多个正常样品的所述扩增子确定的多个经归一化读段计数的中值。所述检测所述基因融合的步骤可以进一步包含通过应用威尔科克森秩检验将多个所述二元分割评分和与对照基因的第二多个扩增子相对应的第二多个二元分割评分进行比较来确定p值。所述检测所述基因融合的步骤可以进一步包含对所述p值应用阈值。所述检测所述基因融合的步骤可以进一步包含对所述不平衡评分应用阈值。
[0181]
根据示例性实施例,提供了一种用于检测基因融合的方法,所述方法包括:(a)在引物池存在的情况下对核酸样品进行扩增以产生多个扩增子,所述引物池包含靶向驱动基因的多个外显子
‑
外显子连接处的引物,其中所述扩增子与所述外显子
‑
外显子连接处相对应;(b)对所述扩增子进行测序以产生多个读段;(c)将所述读段与参考序列进行比对;(d)通过将与每个扩增子相对应的读段数量除以所述驱动基因的所述扩增子之中的最大读段数量来对读段数量进行归一化以得到每个扩增子的经归一化读段计数;(e)对所述驱动基
因的所述扩增子的所述经归一化读段计数应用基线校正以形成经校正读段计数,其中所述扩增子的所述经校正读段计数通过所述经归一化读段计数除以所述扩增子的基线值的log2确定;(f)计算每个经校正读段计数的二元分割评分以提供与所述多个扩增子相对应的多个二元分割评分;以及(g)基于与最大绝对二元分割评分相对应的扩增子指数来确定所述基因融合的预测断点。所述计算二元分割评分的步骤可以进一步包含:计算从第一扩增子到第i扩增子的经校正读段计数x的部分和s
i
,其中s
i
=x1+
…
+x
i
;以及计算从所述第一扩增子到第n扩增子的所有经校正读段计数的总和s
n
,其中s
n
=x1+
…
+x
n
,其中n是经校正读段计数的总数。所述计算二元分割评分的步骤可以进一步包含通过以下确定第i扩增子的二元分割评分:
[0182][0183]
所述方法可以进一步包含基于观察到的不平衡值和预期不平衡值之比来确定不平衡评分。预期不平衡值可以基于经基线归一化读段计数的阵列,其中所述阵列中的阵列元素的数量是n,并且其中经基线归一化读段计数与正常样品的扩增子相对应。所述确定不平衡评分的步骤可以进一步包含:(a)计算所述阵列的从阵列元素(1+b)到阵列元素n的所述经基线归一化读段计数的第一总和,其中b为预测断点;计算所述阵列的从阵列元素1到所述阵列元素n的所述经基线归一化读段计数的第二总和;以及将所述第一总和除以所述第二总和以形成所述预期不平衡值。所述观察到的不平衡值可以基于所述经归一化读段计数的阵列,其中所述阵列中的阵列元素的数量为n。所述确定不平衡评分的步骤可以进一步包含:计算所述阵列的从阵列元素(1+b)到阵列元素n的所述经归一化读段计数的第一总和,其中b为所述预测断点;计算所述阵列的从阵列元素1到所述阵列元素n的所述经归一化读段计数的第二总和;以及将所述第一总和除以所述第二总和以形成所述观察到的不平衡值。所述方法可以进一步包含通过计算多个正常样品中的对应扩增子的多个经归一化读段计数的中值来确定所述扩增子的所述基线值。所述方法可以进一步包含通过应用威尔科克森秩检验将所述多个二元分割评分和与对照基因的第二多个扩增子相对应的第二多个二元分割评分进行比较来确定p值。
[0184]
根据示例性实施例,提供了一种用于检测基因融合的系统,所述系统包括机器可读存储器和与所述存储器连通的处理器,其中所述处理器被配置成执行机器可读指令,所述机器可读指令当由所述处理器执行时使所述系统执行方法,所述方法包括:(a)在引物池存在的情况下对核酸样品进行扩增以产生多个扩增子,所述引物池包含靶向驱动基因的多个外显子
‑
外显子连接处的引物,其中所述扩增子与所述外显子
‑
外显子连接处相对应;(b)对所述扩增子进行测序以产生多个读段;(c)将所述读段与参考序列进行比对;(d)通过将与每个扩增子相对应的读段数量除以所述驱动基因的所述扩增子之中的最大读段数量来对读段数量进行归一化以得到每个扩增子的经归一化读段计数;(e)对所述驱动基因的所述扩增子的所述经归一化读段计数应用基线校正以形成经校正读段计数,其中所述扩增子的所述经校正读段计数通过所述经归一化读段计数除以所述扩增子的基线值的log2确定;(f)计算每个经校正读段计数的二元分割评分以提供与所述多个扩增子相对应的多个二元
分割评分;以及(g)基于与最大绝对二元分割评分相对应的扩增子指数来确定所述基因融合的预测断点。所述计算二元分割评分的步骤可以进一步包含:计算从第一扩增子到第i扩增子的经校正读段计数x的部分和s
i
,其中s
i
=x1+
…
+x
i
;以及计算从所述第一扩增子到第n扩增子的所有经校正读段计数的总和s
n
,其中s
n
=x1+
…
+x
n
,其中n是经校正读段计数的总数。所述计算二元分割评分的步骤可以进一步包含通过以下确定第i扩增子的二元分割评分:
[0185][0186]
所述方法可以进一步包含基于观察到的不平衡值和预期不平衡值之比来确定不平衡评分。预期不平衡值可以基于经基线归一化读段计数的阵列,其中所述阵列中的阵列元素的数量是n,并且其中经基线归一化读段计数与正常样品的扩增子相对应。所述确定不平衡评分的步骤可以进一步包含:(a)计算所述阵列的从阵列元素(1+b)到阵列元素n的所述经基线归一化读段计数的第一总和,其中b为预测断点;计算所述阵列的从阵列元素1到所述阵列元素n的所述经基线归一化读段计数的第二总和;以及将所述第一总和除以所述第二总和以形成所述预期不平衡值。所述观察到的不平衡值可以基于所述经归一化读段计数的阵列,其中所述阵列中的阵列元素的数量为n。所述确定不平衡评分的步骤可以进一步包含:计算所述阵列的从阵列元素(1+b)到阵列元素n的所述经归一化读段计数的第一总和,其中b为所述预测断点;计算所述阵列的从阵列元素1到所述阵列元素n的所述经归一化读段计数的第二总和;以及将所述第一总和除以所述第二总和以形成所述观察到的不平衡值。所述方法可以进一步包含通过计算多个正常样品中的对应扩增子的多个经归一化读段计数的中值来确定所述扩增子的所述基线值。所述方法可以进一步包含通过应用威尔科克森秩检验将所述多个二元分割评分和与对照基因的第二多个扩增子相对应的第二多个二元分割评分进行比较来确定p值。
[0187]
根据示例性实施例,提供了一种非暂时性机器可读存储介质,所述非暂时性机器可读存储介质包括指令,所述指令当由处理器执行时使所述处理器执行用于检测基因融合的方法,所述方法包括:(a)在引物池存在的情况下对核酸样品进行扩增以产生多个扩增子,所述引物池包含靶向驱动基因的多个外显子
‑
外显子连接处的引物,其中所述扩增子与所述外显子
‑
外显子连接处相对应;(b)对所述扩增子进行测序以产生多个读段;(c)将所述读段与参考序列进行比对;(d)通过将与每个扩增子相对应的读段数量除以所述驱动基因的所述扩增子之中的最大读段数量来对读段数量进行归一化以得到每个扩增子的经归一化读段计数;(e)对所述驱动基因的所述扩增子的所述经归一化读段计数应用基线校正以形成经校正读段计数,其中所述扩增子的所述经校正读段计数通过所述经归一化读段计数除以所述扩增子的基线值的log2确定;(f)计算每个经校正读段计数的二元分割评分以提供与所述多个扩增子相对应的多个二元分割评分;以及(g)基于与最大绝对二元分割评分相对应的扩增子指数来确定所述基因融合的预测断点。所述计算二元分割评分的步骤可以进一步包含:计算从第一扩增子到第i扩增子的经校正读段计数x的部分和s
i
,其中s
i
=x1+
…
+x
i
;以及计算从所述第一扩增子到第n扩增子的所有经校正读段计数的总和s
n
,其中s
n
=x1+
…
+x
n
,其中n是经校正读段计数的总数。所述计算二元分割评分的步骤可以进一步包含通过以下确定第i扩增子的二元分割评分:
[0188][0189]
所述方法可以进一步包含基于观察到的不平衡值和预期不平衡值之比来确定不平衡评分。预期不平衡值可以基于经基线归一化读段计数的阵列,其中所述阵列中的阵列元素的数量是n,并且其中经基线归一化读段计数与正常样品的扩增子相对应。所述确定不平衡评分的步骤可以进一步包含:(a)计算所述阵列的从阵列元素(1+b)到阵列元素n的所述经基线归一化读段计数的第一总和,其中b为预测断点;计算所述阵列的从阵列元素1到所述阵列元素n的所述经基线归一化读段计数的第二总和;以及将所述第一总和除以所述第二总和以形成所述预期不平衡值。所述观察到的不平衡值可以基于所述经归一化读段计数的阵列,其中所述阵列中的阵列元素的数量为n。所述确定不平衡评分的步骤可以进一步包含:计算所述阵列的从阵列元素(1+b)到阵列元素n的所述经归一化读段计数的第一总和,其中b为所述预测断点;计算所述阵列的从阵列元素1到所述阵列元素n的所述经归一化读段计数的第二总和;以及将所述第一总和除以所述第二总和以形成所述观察到的不平衡值。所述方法可以进一步包含通过计算多个正常样品中的对应扩增子的多个经归一化读段计数的中值来确定所述扩增子的所述基线值。所述方法可以进一步包含通过应用威尔科克森秩检验将所述多个二元分割评分和与对照基因的第二多个扩增子相对应的第二多个二元分割评分进行比较来确定p值。
[0190]
核酸序列数据可以使用包含但不限于以下的各种技巧、平台或技术产生:毛细电泳法、微阵列、基于连接的系统、基于聚合酶的系统、基于杂交的系统、直接或间接核苷酸鉴别系统、焦磷酸测序、基于离子或ph的检测系统、基于电子签名的系统等。
[0191]
核酸测序平台(如核酸测序仪)的各个实施例可以包含如图31的框图中所显示的组件。根据多个实施例,测序仪器1200可以包含流体递送和控制单元1202、样品处理单元1204、信号检测单元1206以及数据采集、分析和控制单元1208。用于下一代测序的仪器、试剂、文库和方法的各种实施例描述于美国专利申请公开案第2009/0127589号和第2009/0026082号中。仪器1200的各种实施例可以提供可以用于并行地,如基本上同时从多个序列收集序列信息的自动化测序。
[0192]
在各个实施例中,流体递送和控制单元1202可以包含试剂递送系统。试剂递送系统可以包含用于各种试剂的存储的试剂储集器。试剂可以包含基于rna的引物、正向/反向dna引物、用于连接测序的寡核苷酸混合物、用于合成测序的核苷酸混合物、任选的ecc寡核苷酸混合物、缓冲剂、洗涤试剂、阻断试剂、汽提试剂等。此外,试剂递送系统可以包含移液系统或连续流动系统,其连接样品处理单元与试剂储集器。
[0193]
在各个实施例中,样品处理单元1204可以包含样品室,如流槽、基板、微阵列、多孔盘等。样品处理单元1204可以包含多个槽道、多个通道、多个孔或其它基本上同时处理多个样品组的构件。此外,样品处理单元可以包含多个样品室以使得能够同时处理多个轮次。在特定实施例中,系统可以对一个样品室执行信号检测,并且基本上同时处理另一样品室。此外,样品处理单元可以包含用于移动或操控样品室的自动化系统。
[0194]
在各个实施例中,信号检测单元1206可以包含成像或检测传感器。举例来说,成像或检测传感器可以包含ccd、cmos、离子或化学传感器(如覆盖cmos或fet的离子敏感层)、电流或电压检测器等。信号检测单元1206可以包含励磁系统以引起探针(如荧光染料)发射信号。励磁系统可以包含照明源,如弧光灯、激光、发光二极管(led)等。在具体实施例中,信号检测单元1206可以包含用于将光从照明源传输到样品或从样品传输到成像或检测传感器的光学系统。或者,信号检测单元1206可以提供基于电子或非光子的检测方法并且因此不包含照明源。在各种实施例中,基于电子的信号检测可以在测序反应期间产生可检测信号或物质时进行。例如,信号可以通过与离子或化学敏感层相互作用的所释放的副产物或部分(例如所释放的离子,如氢离子)的相互作用而产生。在其它实施例中,可检测信号可以由于如用于焦磷酸测序(参见例如美国专利申请公开案第2009/0325145号)中的酶促级联产生,其中焦磷酸酯通过聚合酶的碱基并入产生,所述聚合酶进一步与atp硫酸化酶在腺苷5'磷酰硫酸存在下反应来产生atp,其中所产生的atp可以在荧光素酶介导的反应中耗尽来产生化学发光信号。在另一个实例中,电流的变化可以在核酸穿过纳米孔时,在不需要照明源的情况下检测。
[0195]
在各种实施例中,数据采集分析和控制单元1208可以监测各种系统参数。系统参数可以包含仪器1200的各种部分(如样品处理单元或试剂储集器)的温度、各种试剂的体积、各种系统子组件(如操控器、步进式电机、泵等)的状态,或其任何组合。
[0196]
所属领域的技术人员应了解,仪器1200的各种实施例可以用于实践多种测序方法,包含基于连接的方法、合成测序、单分子方法、纳米孔测序和其它测序技术。
[0197]
在各种实施例中,测序仪器1200可以测定核酸(如聚核苷酸或寡核苷酸)的序列。核酸可以包含dna或rna,并且可以是单股的,如ssdna和rna,或双股的,如dsdna或rna/cdna对。在各个实施例中,核酸可以包含或来源于片段库、配对库、chip片段等。在具体实施例中,测序仪器1200可以从单一核酸分子或从基本上相同的核酸分子的群组获得序列信息。
[0198]
在各个实施例中,测序仪器1200可以按多种不同输出数据文件类型/格式输出核酸测序读取数据,包含(但不限于):*.fasta、*.csfasta、*seq.txt、*qseq.txt、*.fastq、*.sff、*prb.txt、*.sms、*srs和/或*.qv。
[0199]
根据各种例示性实施例,可以使用适当配置和/或编程的硬件和/或软件元件来进行或实施上述教示内容和/或例示性实施例中的任一个或多个的一个或多个特征。确定是否使用硬件和/或软件元件来实施实施例可基于任何数目的因素,例如期望的计算速率、功率水平、耐热性、处理周期预算、输入数据速率、输出数据速率、存储器资源、数据总线速度等,以及其它设计或性能限制。
[0200]
硬件元件的实例可以包含通过以下以通信方式耦合的处理器、微处理器、一个或多个输入设备和/或一个或多个输出装置(i/o)(或外围设备):本地接口电路、电路元件(例如晶体管、电阻器、电容器、电感器等)、集成电路、专用集成电路(asic)、可编程逻辑装置(pld)、数字信号处理器(dsp)、现场可编程门阵列(fpga)、逻辑门、寄存器、半导体装置、芯片、微芯片、芯片组等。本地接口可以包含例如一个或多个总线或其它有线或无线连接、控制器、缓冲器(缓存器)、驱动器、中继器和接收器等,以允许硬件组件之间的适当通信。处理器是用于执行软件,尤其是存储在存储器中的软件的硬件装置。处理器可以是任何定制的或市售的处理器、中央处理单元(cpu)、与计算机相关联的若干处理器中的辅助处理器、基
于半导体的微处理器(例如呈微芯片或芯片组的形式)、宏处理器,或通常用于执行软件指令的任何装置。处理器还可以表示分布式处理架构。i/o设备可以包含输入设备,例如键盘、鼠标、扫描仪、麦克风、触摸屏、用于各种医疗设备和/或实验室仪器的接口、条形码读段器、触控笔、激光读段器、射频装置读段器等。此外,i/o设备还可以包含输出设备,例如打印机、条形码打印机、显示器等。最后,i/o设备还可以包含以输入和输出的形式连通的设备,例如调制器/解调器(调制解调器;用于接入另一个装置、系统或网络)、射频(rf)或其它收发器、电话接口、网桥、路由器等。
[0201]
软件的实例可以包含软件组件、程序、应用、计算机程序、应用程序、系统程序、机器程序、操作系统软件、中间件、固件、软件模块、例程、子例程、函数、方法、操作步骤、软件接口、应用程序接口(api)、指令集、计算代码、计算机代码、代码段、计算机代码段、字、值、符号或其任何组合。在存储器中的软件可以包含一个或多个独立程序,其可以包含用于执行逻辑功能的可执行指令的有序列表。在存储器中的软件可以包含用于识别根据本发明的教示内容的数据流的系统和任何适合的定制或可商购的操作系统(o/s),其可控制例如系统等其它计算机程序的执行,并且提供排程、输入
‑
输出控制、文件和数据管理、存储器管理、通信控制等。
[0202]
根据各种示例性实施例,可使用可存储指令或指令集的适当地配置和/或编程的非暂时性机器可读介质或物件来执行或实施上述教示内容和/或示例性实施例中的任一个或多个的一个或多个特征,所述指令或指令集如果由机器执行,那么可使机器执行根据示例性实施例的方法和/或操作。此类机器可以包含例如任何合适的处理平台、计算平台、计算装置、处理装置、计算系统、处理系统、计算机、处理器、科学或实验室仪器等,并且可使用硬件和/或软件的任何合适的组合来实施。机器可读介质或物件可以包含例如任何合适类型的存储器单元、存储器装置、存储器物件、存储器介质、存储装置、存储物件、存储介质和/或存储单元,例如存储器、可移动介质或不可移动介质、可擦除介质或不可擦除介质、可写或可重写介质、数字或模拟介质、硬盘、软盘、只读存储器光盘(cd
‑
rom)、可刻录光盘(cd
‑
r)、可重写光盘(cd
‑
rw)、光盘、磁性介质、磁光介质、可移动存储卡或盘、各种类型的数字多功能光盘(dvd)、磁带、磁带盒等,包含适用于计算机的任何介质。存储器可以包含易失性存储器元件(例如随机存取存储器(ram,如dram、sram、sdram等))和非易失性存储器元件(例如rom、eprom、eerom、闪存储器、硬盘驱动器、磁带、cdrom等)中的任一个或组合。此外,存储器可并入电子、磁性、光学和/或其它类型的存储介质。存储器可以具有分布式结构,其中各种组件彼此远离地定位,但仍通过处理器接入。指令可以包含使用任何适合的高阶、低阶、面向对象、视觉、经编译和/或经解译的编程语言实施的任何合适类型的代码,例如源代码、经编译的代码、解译的代码、可执行码、静态代码、动态代码、加密的代码等。
[0203]
根据各种示例性实施例,可至少部分地使用分布式、丛集、远程或云计算资源来执行或实施上述教示内容和/或示例性实施例中的任一个或多个的一个或多个特征。
[0204]
根据各种示例性实施例,上述教示内容和/或示例性实施例中的任一个或多个的一个或多个特征可使用源程序、可执行程序(靶代码)、脚本或任何其它包括待执行的指令集的实体来执行或实施。在源程序情况下,所述程序可以通过可以包含或不包含在存储器中的编译器、汇编器、解释器等翻译以便与o/s一起正确地操作。指令可以使用以下来书写:(a)具有数据类和方法类的面向对象的编程语言;或(b)具有例程、子例程和/或函数的过程
编程语言,可以包含例如c、c++、r、pascal、basic、fortran、cobol、perl、java和ada。
[0205]
根据各种例示性实施例,上述例示性实施例中的一个或多个可以包含向用户接口装置、计算机可读存储介质、本地计算机系统或远程计算机系统发送、显示、存储、打印或输出与可以通过这类例示性实施例生成、访问或使用的任何信息、信号、数据和/或中间结果或最终结果有关的信息。例如,这类发送、显示、存储、打印或输出的信息可以采用可搜索和/或可过滤的运行和报告、图片、表格、图表、图形、电子表格、相关性、序列和其组合列表的形式。
[0206]
本领域技术人员依据上述描述中可以理解,本教导可以以多种形式实施,并且各个实施例可以单独或组合实施。因此,虽然已经结合其特定实例描述了本教导的实施例,但是本教导的实施例和/或方法的真实范围不应如此进行限制,因为在研究附图、说明书和以下权利要求时,其它修改对于熟练的从业者来说将变得显而易见。
上一篇:新产业:公司获得发明专利证书
下一篇:Bioss 34种标签抗体打开您的方便之门