一口气发布6篇Nature、15篇子刊,史上最大规模揭
这一次,科学界以前所未有的规模,揭秘了癌症的复杂性。
规模有多大?Nature一口气发布6篇论文(一篇封面),子刊发布15篇论文。

Nature的这6项研究来自全基因组泛癌分析(Pan-Cancer Analysis of Whole Genomes, PCAWG)联盟,这是他们迄今为止最为全面的癌症基因组荟萃分析:
涵盖38种肿瘤的2658个癌症基因组进行了测序和分析。
以往的研究主要集中在癌症基因组的蛋白编码区域,而此次是分析了整个基因组。
这6篇论文,每篇都从不同方面研究了癌症遗传学,这对于理解癌症的完整遗传复杂性至关重要。
正如Nature评论称:
癌症和云基因组学里的一个里程碑
那么,接下来量子便带大家对6篇论文进行一一解读。
全基因组泛癌分析登上Nature封面
封面文章:Pan-cancer analysis of whole genomes
封面论文汇总信息,概述了PCAWG数据集的广度和深度。
研究人员报告了38个肿瘤类型中,2658个泛癌基因组及其匹配的正常组织的综合分析,其中包括2605个原发肿瘤和173个转移灶或局部复发。
研究数据来自1469名男性(55%)和1189名女性(45%),平均年龄为56岁(范围为1-90岁)。
平均而言,每个癌症基因组均携带4-5个驱动突变(driver mutations),从而为癌细胞提供了选择性优势。
研究涵盖的38种肿瘤之中,仅有5%没有发现驱动突变。
许多癌症表现出了被称作染色体异常(占肿瘤17.8%)和染色体脱色(占22.3%)的基因组突变标志,这导致了基因组的主要结构发生变化。
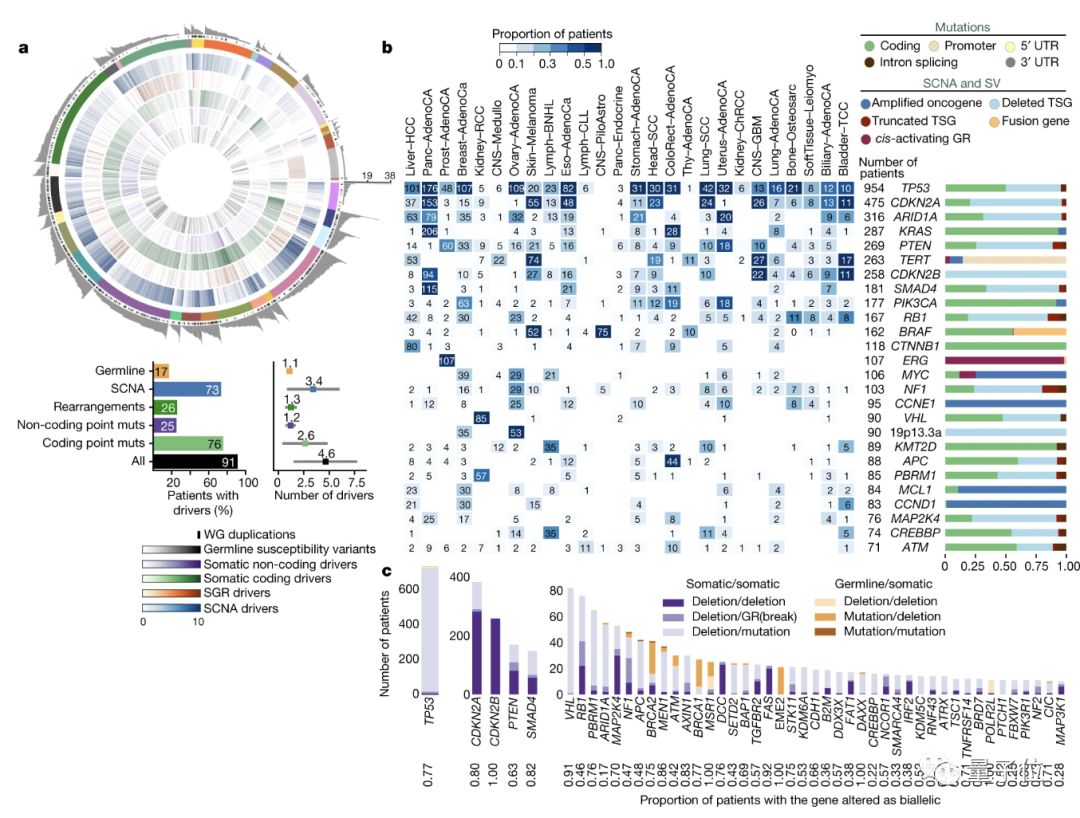
这也就意味着,大规模结构突变在癌症当中发挥着广泛的作用。
研究数据可供全球癌症研究人员下载(地址见文末)。
Analyses of non-coding somatic drivers in 2,658 cancer whole genomes
在这项研究中,研究人员着手确定非编码DNA中的遗传驱动因子。
准确检测非编码区突变要比检测编码区突变难度更大,因此研究人员提出了两种驱动突变发现方法。
研究揭示了新的非编码区驱动突变,比如关键肿瘤抑制基因TP53的5‘端非编码区中发生的复杂突变,FKBIZ和TOB1的3’端非编码区中发生的突变,BRD4的局灶性缺失,以及AKR1C基因位点的重排。
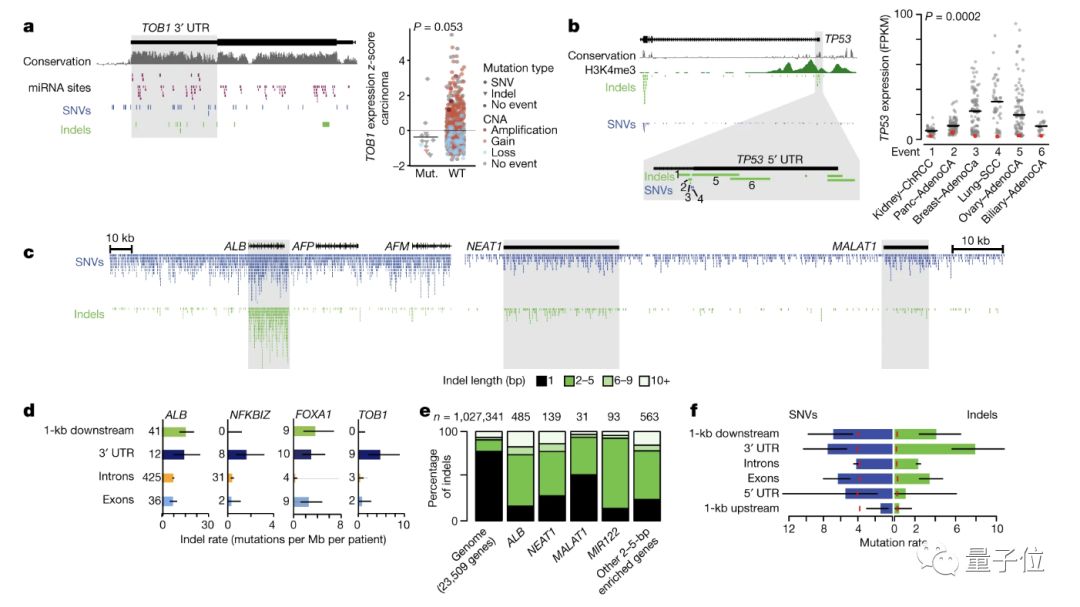
同时,检测结果也对过去发现的个别突变提出了质疑,比如长链非编码的RNA NEAT1和MALAT1。
另外,端粒酶基因TERT的非编码区中相对频繁的突变导致了端粒酶的过表达,这会加助肿瘤细胞不受控制的分裂。
在第三篇和第四篇论文中,重点放在了叫做标签(signature)的基因组畸变。这里的标签是指独特性的DNA序列或单核苷酸位点。
诸如缺陷性的DNA修复机制,或者暴露于环境诱变剂,都会产生特征性的DNA畸变模式。
若是要完善已知的突变标签并发现新的突变标签,那么更大规模的基因组数据集就非常重要。
第三篇论文来自Ludmil B. Alexandrov团队:
The repertoire of mutational signatures in human cancer
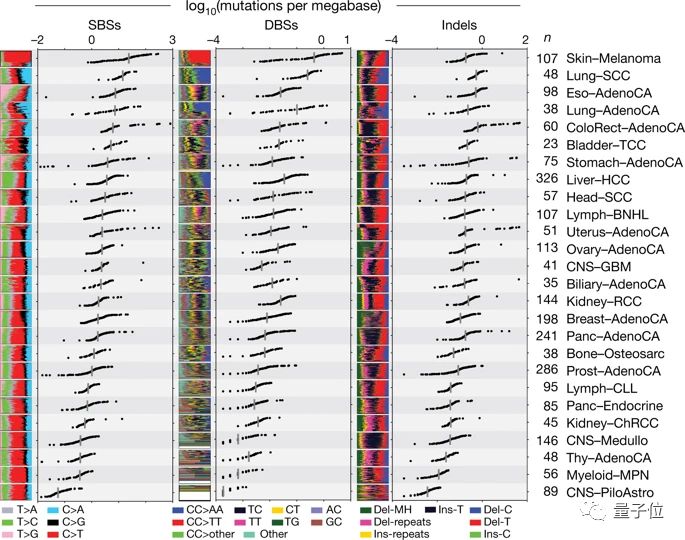
在这项工作中,研究人员使用来自4645个全基因组的84729690个体细胞突变和包含大多数癌症类型的19184个外显子序列来表征突变特征,确定了49个单碱基替换,11个双碱基替换,4个聚类碱基替换和17个小的插入和删除签名。
与以前的分析相比,由于数据集的巨大规模使研究人员能够发现新的签名,分离重叠的签名,并将签名分解为可能代表相关但不同的DNA损伤、修复和/或复制机制的组件。
通过估计每个签名对单个癌症基因组突变目录的贡献,我们揭示了签名与外源性或内源性暴露以及缺陷DNA维持过程的关联。
第四篇论文是来自Yilong Li团队:
Patterns of somatic structural variation in human cancer genomes
上一篇:【新三板报告】2016年新三板生物医药行业研究报告
下一篇:研究显示急性淋巴细胞白血病注定会复发的细胞